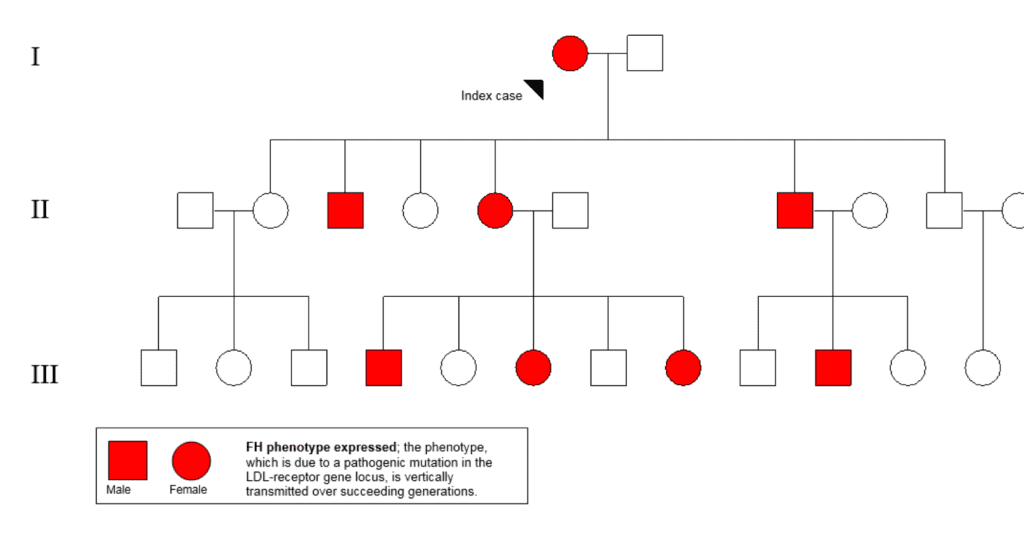
Introduction Familial hypercholesterolaemia (FH) is the most common and serious form of inherited high blood cholesterol. The condition is the result of genetically determined inadequate clearance of low-density lipoprotein (LDL)-cholesterol by the liver. Gene variants that impair this clearance pathway include LDLR (LDL receptor, which mediates the endocytosis of LDL), APOB (apolipoprotein B-100, the receptor ligand) and PCSK9 (proprotein convertase subtilisin/kexin type 9. This enzyme regulates cell surface receptors and degrades LDLR). Inheritance is autosomal dominant, indicating that each first-degree relatives of individuals with heterozygous FH (heFH) have a 1 in 2 chance of having the condition. An example of a pedigree depicting the dominant inheritance in a family with heFH is shown in Fig. 1.
Owing to high LDL-cholesterol levels from birth, individuals with FH are at very high risk of premature coronary artery disease (CAD) particularly if a diagnosis is overlooked and treatment is not initiated early. With an estimated prevalence1 of 1 in 250 and 30 million affected people worldwide, FH is clearly a public health problem. However, it remains under-diagnosed and under-treated.2,3
Screening and diagnosis
There are several screening strategies for detecting FH. These include selective, opportunistic, systematic and universal screening (Box 1). Selective screening of patients with premature CAD, in high risk domains (eg. coronary care units), offers a high detection rate of up to 1 in 10,4 but this strategy does not allow for primary prevention. Recent data has shown that screening via biobanks5 and blood donors6 are good opportunistic approaches. Screening of school children is another option but uptake may be low.7
In addition to opportunistic detection of cases, screening in the community can employ extraction tools that systematically search electronic health records in general practice,8-11 as well as the alerting of an LDL-cholesterol >5.0mmol/l on pathology reports to general practitioners for further assessment of FH.12 These methods, combined with cascade testing of first-and second-degree family members can increase diagnostic yield (Fig.1). Cascade screening or family tracing is a systematic approach for identifying relatives of of an established index case. Although demonstrated to be cost-effective,13,14 cascade screening ries on a supply of new index cases and motivated family members.15 Universal screening of children coupled with the “reverse” cascade testing of parents is potentially an effective method for detecting FH in the community and prior to CAD development.16 This approach appears acceptable17 and cost-effective.18. However, universal screening has only been implemented in Slovenia,19 a country of only 2m people, and requires further evaluation and adaptation to different settings.
Comprehensive guidelines and models of care have been published,20,21 emphasising the importance of integrating screening strategies with clinical care services. Nevertheless, the practicalities and cost-effectiveness of implementing and integrating all of these approaches remains to be demonstrated.
Diagnosis
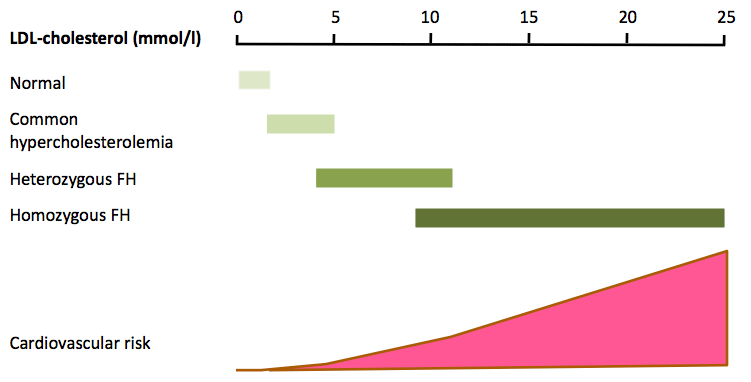
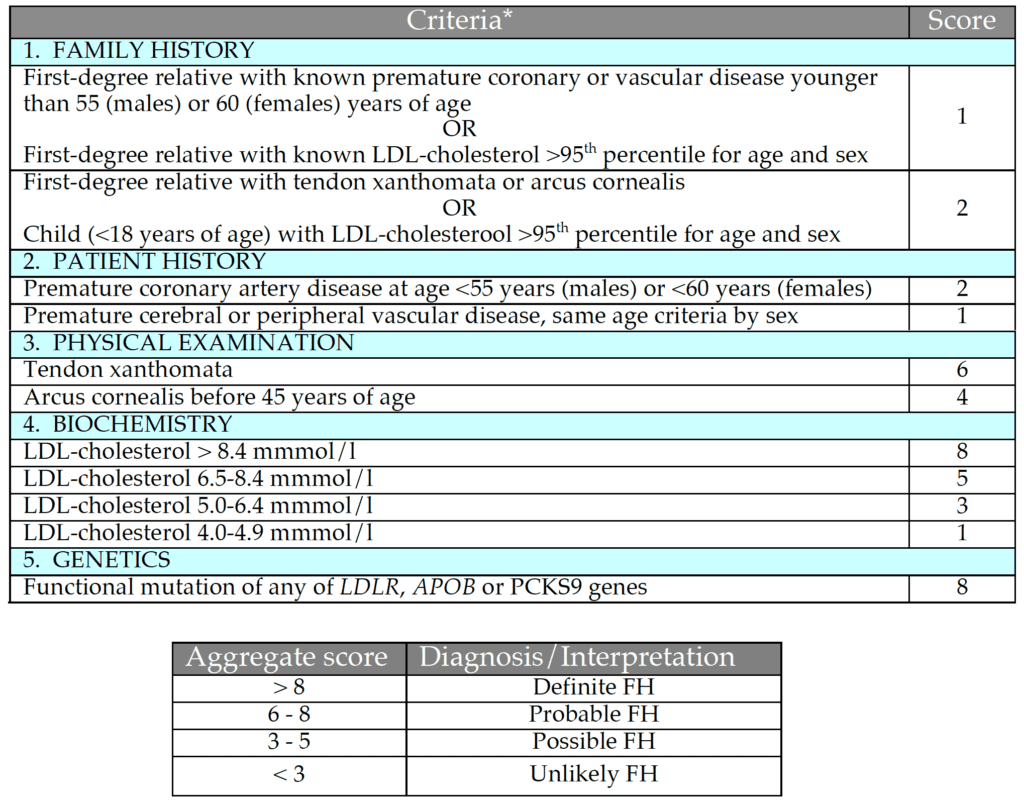
The diagnosis of FH relies primarily on demonstrating a raised untreated plasma LDL-cholesterol concentration, though there is overlap between patients with common hyper-cholesterolaemia and heFH (Fig. 2). Phenotypic tools with additional criteria such as family and personal history of premature CAD and clinical signs such as arcus cornealis and tendon xanthomata are widely used. The most widely used tool is the Dutch Lipid Clinic Network Score (DLCNS) (Table 1)3. US guidelines have simply defined FH in adults using a family history of premature CAD and a plasma LDL-cholesterol greater than 4.9 mmol/l,22,23 but concordance with the “gold standard” genetic diagnosis of FH may be poor.24 An untreated total cholesterol or LDL-cholesterol alone can be used to diagnose FH in first-degree family members once a firm diagnosis has been established in the index case. Cholesterol cut-offs should be adjusted for age and gender.25 There is also an overlap in plasma LDL-cholesterol between patients with one [heFH] or two mutations [homozygous FH (hoFH)] that impair the LDL-cholesterol clearance pathway.26 Recognising the continuum between heFH and hoFH, a pragmatic definition of “severe FH” has been proposed: plasma LDL-cholesterol >10.0 mmol/l alone, >8.0 mmol/l plus one major cardiovascular risk factor or >5.0 mmol/l plus two major cardiovascular risk factors.27 This definition has recently been shown to predict an adverse coronary outcome in the UK.28 However, it lacks the precision of a genetic diagnosis. The use of genetic testing provides an accurate diagnosis of heFH and hoFH, and is therefore important for risk stratification, cost-effective deployment of lipid-lowering therapies and cascade family testing. However, genetic testing has several barriers, such as significant cost, variant interpretations, potential impact on life insurance and limited genetic counselling services.29 Genetic testing is not widely available, but as we rapidly approach a new era of next-generation sequencing30 it should become more widespread. In these circumstances, genetic counselling services for families with FH will be essential.
Assessing FH-related risk
Estimating risk in FH is important to guide therapy, particularly for those that can benefit from primary prevention. Traditional risk factors are known to influence CAD risk in FH,31 but Framingham risk equations do not apply to individuals with FH. FH-specific risk prediction algorithms have been developed.32 These models, however, still require verification across broad sample populations.
Genetic risk factors, such as increased plasma lipoprotein(a) [Lp(a)] concentration,33 a high genetic CAD risk score or high polygenic cholesterol score,34,35 can predict adverse CAD risk, particularly in genetically defined FH. However, the extent to which these genetic scores extend to different populations remains to be determined, as does their clinical use.
Imaging of pre-clinical atherosclerosis may be a valuable tool for assessing risk in FH. Cardiac computed tomography (CT) reliably ascertains coronary artery calcium (CAC) burden and focal stenosis. CAC score has been shown to predict future CAD events in pre-symptomatic FH patients treated with statins. CT angiography also has clinical appeal as it can improve risk stratification, guide treatment intensity and influence adherence to treatment, including lifestyle therapies.37 However, its economic value remains to be demonstrated.
Management
Treatment of elevated LDL-cholesterol in FH involves dietary and lifestyle changes (for example, a heart healthy diet and avoidance of smoking) and pharmacotherapy. Dietary and lifestyle management may be the only treatment option for certain groups, such as pregnant women and younger children.
Drugs
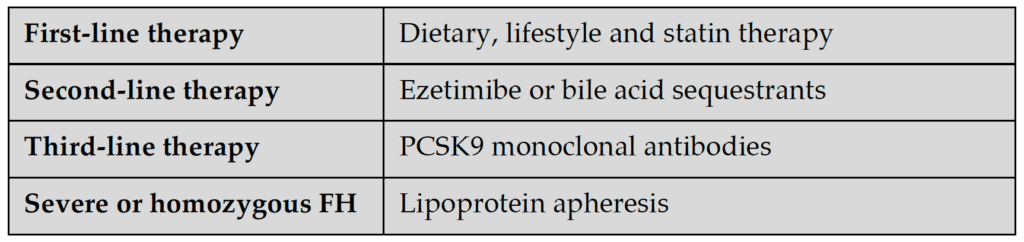
Statins are the first-line pharmacotherapy and is now further supported by new evidence from many cohort and surrogate-endpoint studies as well as clinical trials.38-43 However, a significant proportion of patients do not reach LDL-cholesterol targets on high-intensity statins alone44 and additional therapy with ezetimibe or bile-acid sequestrants may be required.45 Supported by recent clinical trial data, PCSK9 monoclonal antibodies are now recommended as third-line treatment for FH (Table 2).23, 46-53 These antibodies inhibit PCSK9, a convertase enzyme involved in the degradation of LDL receptors in the liver, thereby reducing degradation of LDL receptors and increasing the clearance of LDL cholesterol from the circulation. Given that the majority of adult FH patients will be on more than one drug, interactions and toxicity need to be closely monitored, with appropriate investigations of renal, liver and glucose biochemistry.21
Cholesterol Targets
Guideline recommended treatment targets for FH are a 50% initial reduction in plasma LDL-cholesterol concentration,23 followed by an LDL target of <2.6 mmol/l (if no CAD or major risk factors are present) or <1.8 mmol/l (if CAD or major risk factors are present).2,21,54 More recent guidelines have recommended LDL-cholesterol of <1.8 mmol/l for primary prevention and <1.4 mmol/l for secondary prevention (or very-high risk patients),55,56 which may translate to all FH patients requiring third-line PCSK9 inhibitor therapy. However, the cost or cost-effectiveness implications of these recommendations have yet to be evaluated.
Pregnancy and Breastfeeding
Statins are contraindicated in pregnancy and during breastfeeding,57 although no association has been reported in cohort studies for birth defects, preterm delivery or lower birth weights.58 PCSK9 monoclonal antibodies are also contraindicated in pregnancy.54 Bile acid sequestrants are the only safe drugs to help control FH during pregnancy.21
Children
There is no good evidence for the age of initiation of statins or for LDL-cholesterol treatment targets in children, although Mendelian randomisation studies59 suggest that cholesterol should be lowered as early as possible. Experts recommend that statins should be initiated in heFH children by the age of 10 years, or earlier if preferred by their parents.60,61 Children with hoFH should be immediately treated with statins and additional therapies at the time of diagnosis. Recommended targets are a 50% reduction in plasma LDL-cholesterol concentration60 or LDL-cholesterol <3.5 mmol/l for heFH,61 and <2.6 mmol/l62 and <1.8 mmol/l for hoFH children without and with symptomatic CAD, respectively.62,63
Lipoprotein Apheresis
Lipoprotein apheresis (LA) is recommended for hoFH and severe heFH patients who
remain above LDL-cholesterol treatment targets despite being on maximal drug therapy.21,64 Recent studies confirm that the cardiovascular benefits of LA are related to the degree of reduction in the cumulative exposure to LDL-cholesterol65 and the reduction in arterial inflammation.66 However, LA is not widely available.67 As a final option, liver transplantation may be considered for young patients with hoFH and rapidly progressing CAD.68
New therapies Several new therapies for lowering LDL-cholesterol are in the early phases of clinical trials. These agents could have indications for FH patients who cannot tolerate maximal doses of currently available pharmacotherapy or who have refractory hoFH. PCSK9 may be targeted with long-acting small interfering RNA (inclisiran)69 with much lower injection frequency compared with the PCSK9 monoclonal antibodies. For hoFH patients, therapies that act via a different pathway from current agents may be most useful. One such agent is an ANGPTL3 inhibitor (evinacumab). Evinacumab offers a new and promising opportunity for the further lowering of LDL-cholesterol.70
- Selective screening of high-risk coronary patients
- Opportunistic screening for family history and lipid profile
- Opportunistic alerts and interpretative comments on pathology reports
- Systematic searching of medical records
- Universal screening of children
- “Cascade” screening of family members
Treatment adherence
Poor treatment adherence is associated with increased risk of CAD.71 Hence, it is vital to carefully address concerns and beliefs that patients may have in relation to use of medication.72 This includes health literacy73 as well as the impact of media reports on the perceptions of statins.74 Improving the quality of the shared decision making process75 can be facilitated by the use of decision aids.76
Australian and international initiatives
Developments in Australia include a seminal model of care,20 which has informed international clinical practice on the care of FH.21 A national web-based registry was established in 2015 and contains over 40 clinical sites and more than 1500 subjects across states.77 The Australian Pharmaceutical Benefits Scheme has subsidized PCSK9 monoclonal antibodies for hoFH and heFH patients with and without CAD. In mid-2019 the Medical Services Advisory Committee of the Australian Government recommended that genetic testing for FH be included in the Medicare Benefits Schedule, and final approval is awaited. International efforts have also been initiated to share knowledge and expertise, develop research collaborations and promote early diagnosis and more effective treatments to address this global health challenge.
The drive to improve the detection and management of FH is underscored by several international and regional initiatives. The Familial Hypercholesterolemia Studies Collaboration (FHSC) and the Homozygous International Clinical Collaborators (a registry of hoFH patients), are examples of international efforts to capture a global dataset on FH.78,79 Regionally one can mention the FH Foundation in the US,80 the “10 Countries Study” in the Asia-Pacific region,81 the ScreenPro FH Project in central, southern and eastern Europe,82 the Ibero-American FH Network, and the Gulf FH Foundation. A scientific statement from the American Heart Association has identified a number of key research gaps in FH.22 An ongoing research program focusing on critical questions across the continuum of care is essential21 and these healthcare gaps need to be closed through adequately funded research and implementation.21,22
Patient support and advocacy groups are essential for raising FH awareness in the general public, as well as for advocating for improvement in care and in particular, access to new therapies.83 FH Awareness Day (24th September annually) has been adopted as an annual campaign for FH awareness-raising activities. The impact of patient support networks are exemplified by the activities of the US FH Foundation, Heart UK and the European FH Patient Network.83
Conclusion
Patients with FH have a very high risk of premature CAD and need to be targeted for early intervention. As there is a major shortfall in the detection and treatment of FH worldwide, FH is a global health priority.81 To improve outcomes of FH patients over their lifespan, close collaborations between various stakeholders such as patients, providers, organisations, politicians and the community are essential to translate advances in knowledge into societal-wide health policy and routine high quality care. Updating the WHO Report from 1998 in collaboration with the World Heart Federation is a significant first step for effectively implementing improved global care for FH.84 And the message to individuals is this: if you have a family history of early heart disease please see you doctor and have your cholesterol measured. This may save you and your relatives from a possibly fatal heart attack.
Correspondence: Professor G F Watts, GPO Box X2213 Perth WA 6847 Australia.
Email: gerald.watts@uwa.edu.au








