Introduction
Adult-onset Still’s Disease (AOSD) is a rare inflammatory disease with an estimated prevalence of 1 in 100,0001,2 and an annual incidence of 0.16 to 0.40 per 100,000 depending on the population studied.3,4,5 The underlying pathophysiology remains unclear. However, factors including an imbalance in innate and adaptive immunity geared towards a CD4-Th1 cell response and elaboration of associated inflammatory cytokines including tumour necrosis factor-alpha (TNF-α), interleukin (IL)-1 and IL-6 are thought to contribute to disease development and lend weight to an auto-inflammatory hypothesis.6 Several infectious triggers including viruses (Parvovirus B19, Epstein Barr virus, Cytomegalovirus) and bacteria (Yersinia and Mycoplasma), as well as genetic factors related to HLA linkage have also been implicated.7,8 Some patients who meet classification criteria may have a subacute or chronic unidentified viral infection that may take a different trajectory in respect to recovery compared to auto-inflammatory AOSD.
The classic presentation is a triad of a salmon-coloured evanescent maculopapular rash, high-spiking daily fevers above 39°C and arthralgia or arthritis. In 1992, Yamaguchi et al published classification criteria comprised of major and minor criteria with a diagnosis confirmed if 5 or more criteria were met, of which 2 must be major, and provided infections, malignancy (eg. lymphoma) and other rheumatic diseases (mainly systemic vasculitides) were excluded. The sensitivity and specificity of these criteria were 96.2% and 92.1%, respectively.9 Lack of a standardized algorithm to address the exclusion criteria makes application of these classification criteria in clinical practice or research challenging at times.10
In 2002, a revised classification criteria was published by Fautrel et al which incorporated glycosylated ferritin as a major criterion.11 Due to limited availability of laboratory testing for glycosylated ferritin, the Fautrel criteria has not been used as extensively. Until 1987, AOSD was thought to be a benign condition. However, a study of 21 patients with AOSD by Cush et al revealed different clinical patterns of disease activity (for example, monocyclic or polycyclic patterns, or a chronic non-erosive articular pattern), each associated with different prognoses.12
The hallmark of AOSD pathophysiology is neutrophil and macrophage activation. Multiple cytokines have been implicated viz: IL-1, IL-6, IL-17, IL-18 and TNF-α with treatments aimed at suppressing these pro-inflammatory cytokines in general with non-steroidal anti-inflammatories (NSAIDs), corticosteroids (CS) and csDMARDs.1,8,13,14 However, more evidence from systematic and literature reviews of case reports and case series is emerging for the role of bDMARDs that target specific cytokines. These include tocilizumab (IL-6 inhibitor), anakinra and canakinumab (IL-1 antagonists) as well as TNF-α inhibitors with growing evidence pointing towards efficacy of IL-1 and IL-6 inhibitors in refractory patients (including those refractory to TNF-α inhibitors).15-19 An IL-18 receptor antagonist has recently been investigated.20 Management of AOSD largely depends on the severity of the manifestations and presence of complications including reactive hemophagocytic lymphohistiocytosis syndrome (HLH), cardiac complications, acute kidney failure, respiratory distress, hematologic complications, hepatic failure and multi-organ failure.1,15,21
We conducted a retrospective observational study across 3 metropolitan tertiary hospitals in Perth, Western Australia between January 2009 and 2019 to examine the clinical course and outcomes after 12 months of follow up from diagnosis in AOSD patients and, in particular, to evaluate responses to diverse treatments, especially bDMARDs. The available literature contains scant detail as to the important issue of prognosis and long-term outcomes.
Methods
Patients were identified through their hospital records using the ICD-10 code for AOSD at Sir Charles Gairdner, Fiona Stanley and Royal Perth Hospitals. Inclusion criteria were patients over the age of 18 years fulfilling the Yamaguchi criteria and diagnosed between January 2009 and 2019. Patients with an alternative diagnosis were excluded. The cut-off period for the study follow-up was June 1, 2019 for patients diagnosed in January 2019. Data collected were age, gender, ethnicity, year of initial presentation, presenting symptoms, presence or absence of each of the Yamaguchi criteria to confirm the diagnosis, number of flares since first presentation and last recorded visit. Treatments included NSAIDs, corticosteroids, antibiotics, csDMARDs and bDMARDs. Symptom and serologic outcomes (ferritin, C-reactive protein [CRP] and erythrocyte sedimentation rate [ESR]) in response to these agents were recorded. At 12 months, complete remission (CR) was defined as complete resolution of both clinical and serologic manifestations while partial remission (PR) was defined as resolution of either clinical or serologic manifestations but not both. A flare was defined as relapse of symptoms and serologic markers of disease. The lead author reviewed all records for consistency and assigned a Physician Global Assessment (PGA) a priori using a Visual Analogue Scale (VAS) with a score from 1 (death) to 10 (exceptionally good outcome such as sustained clinical remission).
The following statistical analyses were performed: descriptive summaries, based on frequency distributions for categorical data and medians, inter-quartile ranges (IQR), and boxplots for continuous data. Univariate group comparisons include χ2 and Fisher Exact tests, as appropriate, for categorical comparisons, and Mann-Whitney U tests for comparison of continuous outcomes. Statistical analysis was conducted using IBM SPSS version 24.0 (Armonk, NY). P values of <0.05 were considered statistically significant.
The study was conducted in accordance with the Declaration of Helsinki and was approved by the local Human Research Ethics Committee.
Results
Of the 37 records identified with an ICD-10 code for AOSD, 24 patients met the Yamaguchi inclusion criteria. Six were found to have other diagnoses (3 rheumatoid arthritis, 1 polymyalgia rheumatica, 1 Schnitzler’s syndrome, and 1 polyarteritis nodosa proven on biospy). Three patients did not fulfil the criteria, two had diagnoses made at peripheral hospitals significantly limiting adequate data collection, one was diagnosed prior to the study period with no records available at one of the tertiary hospitals while one patient was assigned an incorrect ICD code with no further assessment to permit confirmation or revision of the diagnosis.

The baseline characteristics of the patients included in the analysis and the treatments received are shown in Table 1. Seventy-nine percent of patients were female, and 54% were Caucasian. The median patient age at diagnosis was 42 years. The commonest symptoms at diagnosis were fevers and arthralgia/arthritis. Seventy-five percent had the classic triad of symptoms while 87.5% had both fever and arthralgia/arthritis
Ferritin concentrations were elevated in all patients: 4.1% had a level >500 µg/L, 41.7% had a level >1000 µg/L and 54.2% had a level >10,000 µg/L. Thus, 95.9% of patients had a ferritin concentration of at least 1000 µg/L while 95.8% had a concentration over 1500 µg/L ( >5 times upper limit of normal in our laboratory). Sixty-seven percent of patients requiring CS were commenced on oral prednisolone with a median dose of 50mg daily while 33.3% required intravenous CS before stepping down to oral therapy.
Eight of 18 patients (44%) received at least one csDMARD, while 39% received ≥2 csDMARDs. Of the bDMARD recipients, 2 received tocilizumab and 2 received anakinra with a good response to each. Three patients were started on one agent then switched due to a side effects (1 developed congestive cardiac failure on tocilizumab). Two had an inadequate response.
Outcomes
Factors associated with CR vs PR at 12 months follow up
Thirty-three percent of patients were in CR, while 38% were in PR at 12 months. Seven patients (29%) were excluded from this analysis as 6 had incomplete records. One patient died whilst hospitalized secondary to HLH and multi-organ failure complicating AOSD. Patients with a lower baseline ferritin (median 4905 µg/L) achieved CR more often compared to those with a higher baseline ferritin who tended only to achieve PR (median 11,000 µg/L). However, these differences were not statistically significant (p-value 0.541).
Whether different treatments were associated with CR or PR at 12 months follow-up was evaluated (Table 2). No statistically significant differences were observed between the treatments that patients received and the achievement of CR or PR (p-value 0.645 for CS, 0.792 for csDMARDs, 0.40 for bDMARDs).
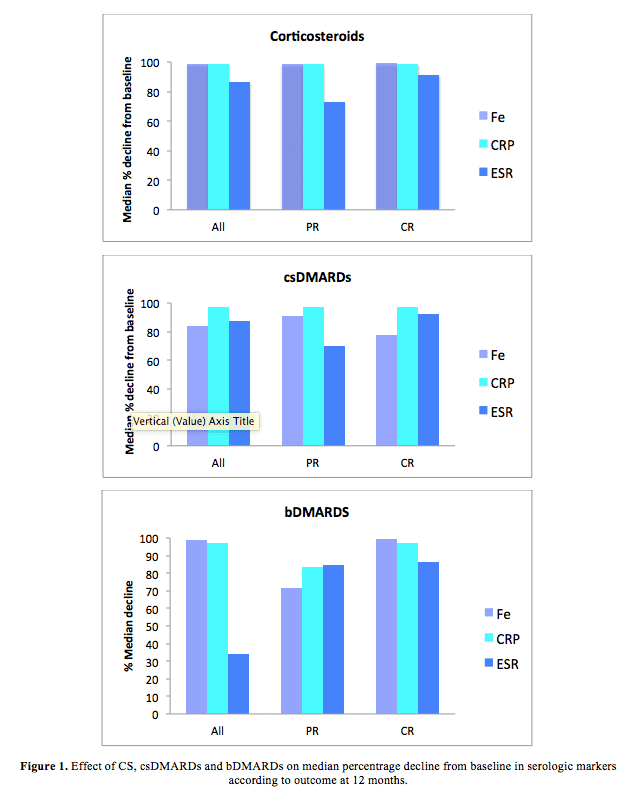
The effect of different treatments on the median percentage reduction from baseline in serologic markers of remission at 12 months follow-up is shown in Figure 1. There was a non-statistically significant greater median percentage reduction from baseline in ferritin concentrations (p-value 0.62) and CRP (p-value 0.681) in the CS group, and for CRP in the csDMARD group (p-value 1.00). There were no significant differences observed in the bDMARD group in terms of serologic markers of outcome except for a significant reduction in ESR in those who achieved PR but this occurred in the patient commenced on tocilizumab (p-values 0.70 for ferritin, 0.85 for CRP, 0.20 for ESR).
Factors affecting AOSD flare rates
The flare rate per 100 patient-years was 36.9 with CS alone, 43.5 with csDMARDs in combination with CS and 40.0 with bDMARDs in combination with over the period of observation.
Figure 2 reveals an apparent inverse trend between flare frequency and baseline ferritin. For example, in patients with a lower baseline ferritin (eg. ≤500 µg/L) 5 of 8 (62.5%) flared, whereas those with higher baseline ferritin concentrations (eg. ≥5000 µg/L) 8 of 15 (53.3%) were observed to flare. These differences were not statistically significant (p-value 0.256).
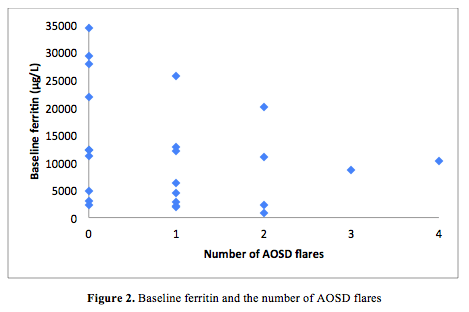
We evaluated whether baseline ferritin predicts the need for csDMARD or bDMARD therapy at 12 months. Although the median baseline ferritin concentration was higher in the csDMARD and bDMARD groups compared to those who received no treatment (2 patients) or corticosteroid monotherapy (1 patient), these differences were not statistically significant (Fig. 1). In those with baseline ferritin greater than 10,000 µg/L, 5 of 11 (45%) experienced 1 or more flares, in contrast to 6 of 9 (66.7%) whose ferritin was relatively low. Of interest, the median baseline ferritin was higher in the group not receiving cs-DMARDs or bDMARDs compared to those receiving these treatments (Fig. 2), but this was also not statistically significant (p = 0.202 for csDMARDs and 0.884 for bDMARDs).

Physician Global Assessment
The PGA assigned at 12 months was examined in relation to the receipt of csDMARDs, bDMARDs or a combination thereof. Higher PGA scores compatible with better outcomes were observed in those who received no treatment or corticosteroids alone (Table 1). Moreover, a greater proportion of patients receiving csDMARDs had a PGA score of ≥7 at 12 months compared to those who were not receiving this therapy. No significant difference in PGA scores was observed amongst those who were receiving bDMARDs (Figure 3). These results were not statistically significant (p-value 0.584 for csDMARDs, 1.00 for bDMARDs).
A trend toward lower PGA scores was observed in patients with a higher baseline ferritin concentration (median ferritin 10,800µg/L for PGA < 7 vs 5,150µg/L for PGA > 7) but this difference was not statistically significant (p-value 0.462).
Discussion
AOSD is a rare multi-system immuno-inflammatory disorder. Ninety six percent of our patients had a ferritin concentration > 1000 µg/L, greater than the frequency of around 70% reported from Japan,22 but identical to that of 95.8% recently reported in a large Chinese series.23 Thus hyperferritinaemia is highly suggestive of the diagnosis of AOSD. Only a few other conditions cause a similar picture, for example sepsis, macrophage activation syndrome, and HLH, all of which can also complicate AOSD. Hyperferritinaemia also occurs in genetic haemochromatosis, but patients are rarely febrile on first presentation. It remains unclear if ferritin is directly involved in disease pathogenesis or is simply a silent bystander, as the same cytokines observed in AOSD also regulate ferritin synthesis at a transcriptional, post-transcriptional and translational stage.24
Given the rarity of the disease, long-term data addressing outcomes in AOSD are understandably scarce. Descriptions of physician-reported outcomes for established treatments are limited. Our study of AOSD patients collected over 10 years was intended to help ascertain which factors might predict remission at 12 months and to assess the effect of treatments on serologic markers of disease and on remission.
In our small cohort for whom adequate data was available for analysis in 17 of the 24 patients, all patients required corticosteroids therapy at the time of diagnosis, due to the inefficacy of NSAIDs, consistent with the literature.13 CsDMARDs were required in 75% of our cohort as a high proportion of patients demonstrated steroid dependency. This was significantly higher than the 45% described in other studies.13,14,21,25 Factors reportedly associated with corticosteroid dependence in other studies include younger age at onset, splenomegaly, higher ESR and lower glycosylated ferritin concentrations. Corticosteroids were found to have greater efficacy in patients with systemic symptoms, as opposed to articular symptoms,13 which raises the possibility that a high proportion of our cohort may eventually develop the chronic articular disease pattern.12 Our results also revealed patients were receiving csDMARDs and bDMARDs (in addition to CS therapy) for a median of up to 11 months. Only 37.5% and 22.2% of csDMARD recipients achieved CR and PR, respectively. Similarly, only 25% and 11.1% of patients receiving bDMARDs achieved CR and PR. This is significantly lower than the reported efficacy rates in the literature. For example, in methotrexate recipients, in whom 70% CR and 88% PR rates have been described.13,14 A 77% CR rate was achieved with anakinra, whereas 86% and 96% improvement in joint and systemic symptoms (respectively) has been described with tocilizumab.26 The main difference between our study and others is that our patient numbers for each treatment group were significantly smaller. The length of follow-up may also be relevant to the differing observations. Nonetheless, we consider that long term results may not be as good as has been previously suspected and suggest that 2- and 5-year outcomes should be collected for additional clarity.
Low ferritin concentrations at presentation correlated weakly with complete remission, implying that low ferritin may be a possible predictor of good recovery. More data is required to validate this hypothesis. More patients with a high baseline ferritin concentration required csDMARDs and bDMARDs compared to those receiving no treatment or CS monotherapy. Fewer patients achieved CR at 12 months follow up, a trend suggesting that higher baseline disease activity, as reflected by the ferritin concentration may be a feature of disease that is more difficult to control or less likely to remit. Patients with higher baseline ferritin concentrations had fewer or no flares. This was unexpected and requires confirmation in a larger cohort.
Treatment with corticosteroids, csDMARDs and bDMARDS resulted in a marked reduction in serologic markers, similar to that found in other studies.15 We found no statistically significant differences between those achieving CR and PR in the treatments received or the specific serologic marker. More patients receiving csDMARDs +/- bDMARDs (including CS) had a higher PGA score at 12 months than those receiving no treatment or corticosteroid monotherapy. We also found an inverse trend towards a higher baseline ferritin and PGA. These findings suggest that higher baseline disease activity more likely predisposes to persistent or refractory disease, which in turn adversely affects patient quality of life. Extended follow-up studies are required to gather more information concerning the sustainability of remissions and to ascertain how frequently joint and other organ damage becomes apparent in the long term.
The strengths of this study include detailed chart reviews in all 37 participants who received ICD-10 coding for AOSD, the rigorous application of internationally accepted classification criteria in combination with long-term follow-up wherever possible to confirm or refute the diagnosis, and the capture of extensive clinical and laboratory data. A further strength is the focus on measures of outcome, for which there is a paucity of information in the medical literature. This included determination of remission rates and PGAs.
The main limitations are the retrospective nature of the study and, importantly, the small sample size, despite the 10-year period over which data was collected. This is consistent with the rarity of AOSD. The low rate of patient retention in tertiary hospital clinics and methods of assessment hindered the ascertainment of long-term outcome data. Regrettably, patient-reported outcomes were not available, and furthermore patient perceptions of treatment efficacy could not be ascertained. Longer term data beyond 12 months was limited to just a few patients, thereby limiting robust statistical analysis.
The acquisition of long term outcome data (eg. at 2 and 5 years) including patient- and physician-reported outcomes should provide greater insight into the extent and duration of remissions on and off treatment. Long-term extension studies should be encouraged when new therapies are investigated in order to gather such outcome data. Finally, interested physicians should be encouraged to establish and contribute to a global AOSD registry and the development of a patient biobank as this could be invaluable for studying pathogenesis and outcomes on a larger scale, across multiple ethnicities and for investigating the role of newer and repurposed therapeutic agents.
In conclusion, AOSD was found to respond less well to treatment than expected. Low complete and partial remission rates were observed and moderately high flare rates were identified over the course of the first 12 months after diagnosis, irrespective of treatment. These findings emphasise the recalcitrant nature of AOSD and in turn, the unmet need for more effective therapy.
Provenance Internally reviewed
Ethical approval: Not required
Conflicts of Interest: None declared
Funding: Not required
Corresponding author: Dr. Pauline Habib, BJC Health, Chatswood, New South Wales 2067, Australia. Email: Paulinehabib@ymail.com








