Introduction
Inclusion body myositis (IBM) is the most commonly acquired skeletal muscle disease associated with aging.1 Its pathogenesis includes both muscle tissue degeneration with protein accumulation (TDP-43, β-amyloid, p62 etc); and autoimmunity with T-cell invasion of non-necrotic muscle fibres. Although it is unclear which of these processes is responsible for disease initiation and remains a source of debate, it is likely that both contribute to muscle degeneration over time. It is known to involve genetic susceptibility factors (HLA-DRB1*0301) that interact with as yet unknown environmental triggers. Men are more commonly affected than women, with a ratio of about 3:1.1,2 The overall prevalence is varied in different populations, and in Western Australia is considered to be 9.3 per million, or 3.5/100,000 in individuals over the age of 50.2,3 The most severely affected muscle groups in IBM include the quadriceps, causing difficulty in rising from low chairs and in climbing stairs, forearm finger flexors causing reduced grip strength, and swallowing muscles causing dysphagia. As the disease progresses, its effect on these muscle groups leads to progressive dependency and disability. Patients usually require the use of a walking aid after about 8 years, and a wheelchair around 10 years. Reduced mobility may also result from secondary functional loss as a result of de-conditioning and reduced lifestyle physical activity or exercise. IBM is poorly responsive to currently available immunosuppressive therapies and its impact on quality of life is considerable.1
Exercise is defined here as a subset of physical activity structured to obtain improvement or maintenance of physical fitness.4 It has long been considered an important part of the therapeutic response in inflammatory myopathies. Exercfise helps prevent loss of muscle mass, allows for maintenance of strength and function, and therefore improves quality of life.5 It is also thought to have an immunomodulatory effect in some neurodegenerative conditions such as multiple sclerosis and autoimmune encephalitis.6 There have been many studies into the effect of exercise in IBM, and most have shown positive results.5,7,8 However, none have combined exercise training and testosterone. This is of clinical importance as it may encourage multimodal management strategies in IBM.
The combination of testosterone and exercise training has been shown to improve upper and lower limb skeletal muscle strength and performance to a greater extent than exercise alone, in healthy young and middle-aged men, and the effects of exercise and testosterone is additive.9 A similar beneficial additive effect of testosterone and exercise on performance without weight gain has been observed in older males with low normal blood testosterone.10 The myotrophic effects of testosterone are dose-dependent in males, and the effect does not wane with age.11 It is not known whether similar beneficial effects would be seen in males with IBM. A study of oxandrolone, a 17alpha-alkylated testosterone analogue, in patients with IBM reported a positive effect of borderline significance on whole-body and upper extremity strength,12 but was limited by differences in pre-intervention muscle power in the two study groups (oxandrolone and placebo) and possible unintended unblinding of treatment allocations. Exercise as a factor was not included in the study. This study is the first to measure the effect of testosterone with or without exercise training in IBM.
This pilot study is a double-blind, randomised, controlled two-arm crossover trial to test the hypothesis that testosterone added to exercise training will improve measures of muscle power and physical activity in men with IBM compared to exercise alone. It was conducted over a 6-month intervention period (12 weeks on each arm with a 2-week washout period), to which was added a 12-month open-label extension (OLE) in response to participant feedback. This manuscript presents the study protocol, approved by the Human Research Ethics Committee at Murdoch University (MU 2017/262), and considers strengths and limitations of the study design.
Methods
Study Design
The study took place over 26 weeks and was split into two treatment periods of 12 weeks, separated by a two-week washout period. After providing informed consent, patients were screened against the inclusion and exclusion criteria (table 1). Exercise was prescribed for the study duration. Participants were randomised to one of two study arms, by allocation of sequentially numbered treatment kits, and received either transdermal testosterone or placebo (consisting of vehicle only) during the first 12-week treatment period, and the alternate treatment during the second 12-week period, after the washout phase. The randomisation was performed offsite by a clinical research pharmacy, which compounded and provided the study medication in a blinded fashion. The pharmacy also retained the randomisation key until after the crossover part of the study was completed.

This was designed as a pilot study, with a target sample size of 20, dictated by the number of participants with IBM that could feasibly be recruited through our myositis clinics in Western Australia. Clinical aspects of the study were undertaken at the Academic Medical Centre, Institute for Immunology and Infectious Diseases (IIID), Murdoch University.
Trial drug details
The testosterone formulation was AndroForte-5® produced by Lawley Pharmaceuticals (Perth, Western Australia). It contains 50 mg/ml of testosterone in an aqueous cream suitable for transdermal application. The recommended treatment regimen in hypogonadal men is 100 mg daily.13 This cream contained testosterone with the addition of cetomacrogol 1000, cetostearyl alcohol, carbopol 940, almond oil, DL-a-tocopheryl acetate (Vitamin E acetate), triethanolamine, Phenonip®, butylated hydroxytoluene, anhydrous citric acid, and purified water. The placebo contained the same ingredients, without testosterone, and had the same application advice. The transdermal cream was applied on an area of the torso with low subcutaneous fat, allowed to dry, and then covered with clothing. To ensure double-blinding, study medication and placebo were supplied in unmarked plain tubes labelled with kit numbers, and provided to participants according to the randomisation schedule.
Participants were provided with drug safety information and education regarding study medication application. They were instructed to return the used medication tubes for drug accountability purposes. Participants recorded study medication application using a paper diary, including location of application (e.g. chest or back).
Exercise training
Exercise training was a combination of light-moderate resistance training, balance training and aerobic exercise; it was personalised according to the stage of disease progression and motor capabilities of each participant. The exercise program was supervised by investigators with at least 10 years experience from the School of Psychology and Exercise Science, Murdoch University (YL and TF). Exercise investigators were blind to treatment allocations.
An individualised progressive home-based exercise program, targeting the whole body, was created to accommodate the large variance in strength and functional capabilities of each participant, and tailored to their functional ability according to baseline assessment outcomes. The duration of each exercise session was designed to be performed for approximately 30-60 minutes for a minimum of three times a week. Participants were prescribed one of three levels with exercise appropriate for individuals with low (for example, wheelchair dependent), moderate (walking aid dependent over long distances), or high (independent walking) mobility. Resistance exercises were conducted using single joint exercises of the lower and upper limbs using elastic bands (Therabands). Multi-joint compound exercises were based on body-weight and focussed on knee extensors and wrist/finger flexors. This was supplemented with aerobic/mobility and functional exercises (i.e. walking, sitting and rising) and balance exercises (i.e. narrow stance, tandem stance). The intensity of the exercise (moderate-light) was determined using the rating of perceived exertion (RPE) Borg scale, with a Borg of 12-14 prescribed.14 A RPE was attained for every exercise prescribed for each participant and this information was used to establish an appropriate intensity and regress or progress the exercise accordingly. In this study Actigraphs (Actigraph, Pensacola, Florida, USA) were trialled for monitoring of exercise, which has been validated in multiple sclerosis trials.15 Fortnightly telephone consultations took place, and during this time, exercise progression (i.e., sets and repetitions) of each exercise were confirmed based on participant feedback and clinical judgement. Example prescription in Week 1 and Week 12 is provided in Table 2. The participants were encouraged to maintain and continue any other physical activity or exercise behaviours that they usually performed.
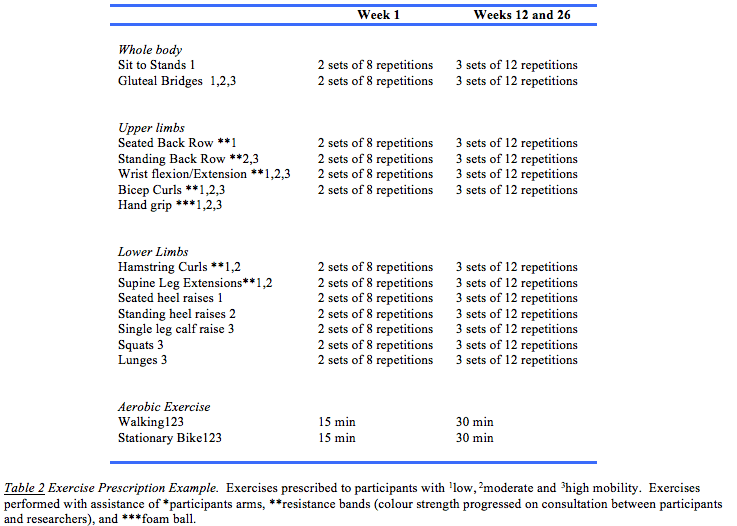
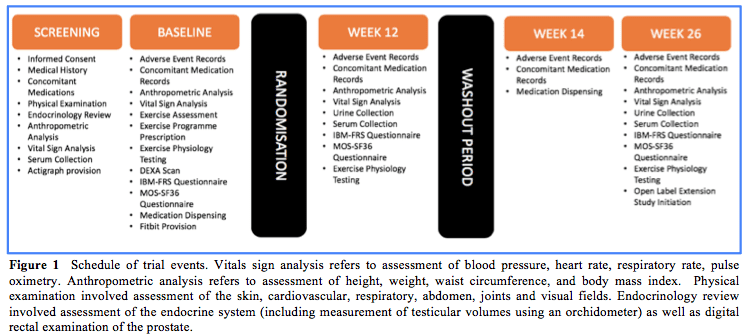
Figure 1 shows a summary of the Schedule of Events for the crossover study. The 12-month Open Label Extension (OLE) study was added to the protocol based on feedback from participants and consultation with the study team, by an ethically approved amendment. This extension was designed to provide additional data on the longer-term safety and tolerability of testosterone in IBM, as well as tracking any further changes in muscle strength over 12 months.
Primary Outcome Measures
The primary outcome measure was improvement or stabilisation of quadriceps isokinetic muscle strength. Quadriceps strength was assessed only the right leg at 30 and 60 degrees from full extension (180 degrees) on a HUMACNORM® isokinetic dynamometer [HumacNORM, Computer Sports Medicine, Stoughton, MA, USA]. Hamstring power was assessed on the same leg and the same knee flexion angles. The order of quadriceps and hamstring tests was kept consistent throughout the study and conducted after a standardised warm-up protocol. Testing was performed at onset and at the end of each treatment period (weeks 0, 12 and 26 respectively, and at 6 and 12-months timepoints during the OLE). All assessors were blind to treatment allocation for assessments conducted within the crossover study period.
A battery of additional functional tasks was conducted to assess strength (Sit-to-stand; hand-grip strength), wak capacity (2-minute walk test; Quantitative gait assessment using a Gaitrite (CIR Systems, Havertown PA, USA) static and dynamic balance tasks (Berg Balance Scale; 4-stage balance test; Functional reach test; Activities-specific balance confidence) as well as a body composition analysis (dual energy x-ray absorptiometry for assessment of lean body mass). Specific questions related to activities of daily living were obtained through two Patient Reported Outcomes (PROs), the 10-point IBM Functional Rating Scale (IBM-FRS) and quality of life through the Short Form Health Survey (SF-36). The IBM-FRS is a functional scale tailored for IBM that is considered a reliable method of assessing disease severity17 and includes questions relating to activites such as dressing. The SF-36 is a measure of eight domains of health utilised to assess limitation in physical activity due to health problems.18 Finally, the participants were asked to record their daily training in the form of a diary. This focussed on the exercises executed, number of repetitions and sets, and duration of exercise. This diary included adverse events reporting, including falls.
Secondary Outcome Measures
1. Engagement in activities monitoring
Exploratory outcome measures included the use of a commercial Fitbit Charge 2 activity tracker (Fitbit, San Francisco, CA, USA), which was also compared to medical-grade actigraphy. Physical activity was objectively measured using a Fitbit applied continuously across the 26-week study period. The Fitbit tracker data gathered a composite of daily measures including number of steps, energy expenditure, floors climbed, minutes of activity and sleep quality. A hip-worn, triaxial GT3X Actigraph accelerometer was also worn, in addition to the Fitbit, during a 7-day period within the first weeks of each 12-week treatment period for the above comparison.
2. Changes in sleep duration and quality
We evaluated if this protocol affected the participants’ sleep patterns using Fitbits worn at night to record sleep duration and quality.
3. Measure of inflammation in biological samples
Serum and urinary inflammatory markers were collected at weeks 0, 12, and 26, and at 6 and 12 months during the OLE, to determine whether testosterone had any additional anti-inflammatory effects compared to exercise alone. These were blood pro-inflammatory cytokine and chemokine markers (IP-10, MCP-1, MIP-1-α, MIP-1β, sICAM-1, GM-CSF, IFN-α, IFN-γ, IL-1α, IL-1β, IL-6, IL-12p70, IL-17A and TNF-α), creatinine kinase activity, luteinising hormone and sex hormones by immunoassay, total testosterone by mass spectrometry, and blood immune cells by flow cytometry. Protein carbonyls in urine were measured at the same time points, as a marker of oxidative stress.
Safety Monitoring
Screening blood tests were performed no more than 28 days prior to inclusion in the study for haemoglobin, haematocrit, creatinine kinase, lipids, testosterone, prostate specific antigen, creatinine, and liver function tests. Safety tests during the study were haemoglobin, haematocrit, lipids and testosterone, measured at weeks 12 and 26, and at 6 and 12 month timepoints during the OLE. At each visit, participants were questioned regarding adverse events using open-ended questioning. Vital signs and physical measurements were collected, including weight and waist measurement. Study compliance was reviewed, including drug accountability.
An adverse event was defined as any unintended consequence, including abnormal serum or urine tests, new symptoms or clinical events. These were recorded and will be assessed for severity and causation. An independent endocrinologist was engaged to provide safety monitoring, and reviewed all safety bloods. Any unexpected adverse events or Serious Adverse Events (SAEs) were reported to MN (as Principal Investigator) and the HREC for review. Participants were free to withdraw from the study at any time without providing a reason.
Statistical Methods
Statistical analysis will be performed using several statistics programmes, including the R statistical programme (https://www.r-project.org ) and GraphPad Prism v 8 for Windows (GraphPad Software, SanDiego, CA USA). Continuous variables will be described by median and 95% confidence intervals. Questionnaires, immunological and exercise data will be analysed using the Wilcoxon Signed Rank test within a matched-paired design. They will be separated into baseline, placebo and treatment groups and analysed with comparisons between treatments (placebo (exercise alone) vs testosterone), and by time (baseline vs treatment and baseline vs placebo
Ethics and Dissemination
This study was approved through the Human Research Ethics Committee at Murdoch University with the approval number 2017/262. The participants were provided with detailed patient information sheets, and had the study explained to them, with the opportunity to ask questions. Written consent was received from all participants on approved informed consent forms prior to undertaking any study activities. Participants were informed of their right to withdraw from the study at any stage without having to provide an explanation.
This study has been registered with the Australian and New Zealand Clinical Trials registry (ANZCTR), registration No. ACTRN12618000755235. Electronic data for the study is stored in computers and servers protected by passwords and firewalls hosted at the Institute for Immunology and Infectious Diseases, Murdoch University. Hard copies will be kept in locked filing cabinets in secure premises. Data collected within Case Report Forms (CRFs), including data stored in the online Electronic Data Collection (EDC) database, will be de-identified. Treatment allocation was unblinded to the research team for data analysis after the completion of the crossover trial period. It is not intended to share individual participant data for the trial with any third party. Results will be made available as per open access requirements. The results will be published regardless of result significance.
Discussion
The publication of the study protocol at this stage of the study provides us with an opportunity to reflect upon strengths and limitations of the study design. Lessons learnt through this study will help us with future design of trials in IBM, and may be applicable to other groups pursuing similar research themes. This study has highlighted a requirement for a focus towards a more homogenous population of individuals with IBM within future trials, so that there is less variability in premorbid functioning. The heterogeneity of the participant group will present challenges for drawing meaningful conclusions from the data, particularly within a small sample size. It is possible, but we feel unlikely, that the study duration (3-month treatment periods within the crossover study), was inadequate to observe a response to exercise and testosterone. The choice of a crossover design was chosen to make best use of the modest number of IBM patients attending our clinic but may have resulted in confounding factors which will be further discussed within the published study results.
The collaborative nature of this investigator-initiated trial has resulted in generation of numerous data sets across the exercise, immunology and clinical teams involved in the study. Whilst appreciating the breadth of information that can be obtained from this data, harmonisation of this data across the study team and data analysis is complex and time-consuming, meaning that results cannot be published and disseminated as swiftly as hoped.
Provenance: Externally reviewed
Funding: This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors. Lawley Pharmaceuticals (West Leederville, Perth, WA 6007) provided study medication and placebo without charge.
Declarations: No conflicts of interest declared
Ethics: Approval obtained from Murdoch University (see text)
Acknowledgements: The authors would like to thank participants and staff who contributed to this study.
Corresponding Author: Dr Sophia Connor, Neurology Department, Royal Perth Hospital, Perth, WA 6001, Australia. Email sophia.connor@health.wa.gov.au.